
A method for efficient calculation of thermal stability of proteins upon point mutations†
 *ac
and
John Z. H.
Zhang
*abde
*ac
and
John Z. H.
Zhang
*abde
Abstract
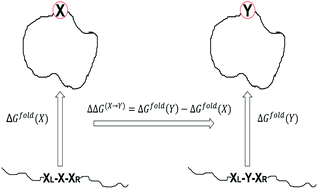
A method for efficient prediction of the relative stability of a protein due to a single amino acid point mutation is presented. In this approach, we calculate the free energy change due to an arbitrary point mutation of a protein from a single MD trajectory of the wild type protein. The method is tested on 27 diverse protein systems with a total of 853 mutations and the calculated relative free energies show a generally good correlation with the experimental values (a correlation coefficient of 0.63). Comparison with the free energy perturbation (FEP) method and the recently developed machine learning methods on two different benchmark data sets shows that the current method is computationally efficient and also numerically reliable for predicting the changes in thermostability upon an arbitrary point mutation of a protein. A discussion is provided on how to further improve the accuracy of the method for the prediction of thermostability of proteins.
- This article is part of the themed collections: Emerging AI Approaches in Physical Chemistry and 2020 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

