
Quantum yields of singlet and triplet chemiexcitation of dimethyl 1,2-dioxetane: ab initio nonadiabatic molecular dynamic simulations†

Abstract
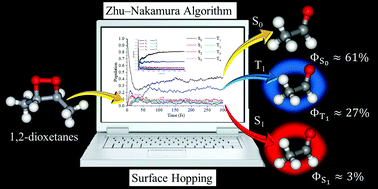
The global nonadiabatic switching on-the-fly trajectory surface hopping simulation at the 8SA-CASSCF quantum level has been performed to estimate the quantum yield of chemiexcitation for the uncatalyzed decomposition reaction of the open-shell biradical trans-3,4-dimethyl-1,2-dioxetane system. The present ab initio nonadiabatic molecular dynamic simulation involving both conical intersection and intersystem crossing is to compute for the first time the population evolution of quantum yields at the four lowest singlet and four lowest triplet states. The simulated results demonstrate not only the stepwise dissociation of O–O and C–C bond breaking, but also confirm the existence of a biradical entropic trap which is responsible for chemiexcitation. The simulated quantum yield of the triplet chemiexcitation ΦT1 = 0.266 ± 0.096 agrees with the experimental value of 0.20 ± 0.04 very well. The present nonadiabatic molecular dynamic simulation of dimethyl 1,2-dioxetanes provides a further advanced understanding and stepping stone for future studies on chemi- and bio-luminescence.
 Please wait while we load your content...
Please wait while we load your content...

