
The interfacial structure and dynamics in a polymer nanocomposite containing small attractive nanoparticles: a full atomistic molecular dynamics simulation study†
 *a
and
Zhong-Yuan
Lu
a
*a
and
Zhong-Yuan
Lu
a
Abstract
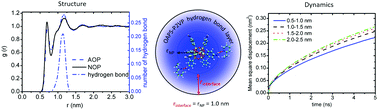
We study the interfacial structure and dynamics of a polymer nanocomposite (PNC) composed of octaaminophenyl polyhedral oligomeric silsesquioxane (OAPS) and poly(2-vinylpyridine) (P2VP) by performing full atomistic molecular dynamics simulations. There are eight aminophenyl groups grafted on the surface of the OAPS particle and the particle has a size comparable to the Kuhn segment of P2VP. These aminophenyl groups can form hydrogen bonds (HBs) with pyridine rings from surrounding P2VP chains. We found that OAPS can form ∼2 HBs on average with surrounding polymer chains. The effect of the HBs is investigated in detail by either switching on or off these HBs in our simulation. By analyzing the interfacial static packing structure and dynamic properties, we demonstrate that the system has an ∼1 nm interface width, similar to the OAPS particle size. We also found that HBs can prevent the further penetration of polymers into the inner zone (grafting layer) of the OAPS, and therefore keep the P2VP chains in the outer layer (>1 nm), remaining bulk-like, which is well consistent with experimental results. In addition, we found that NP diffusion is coupled to the absorbed polymer chains, which also dramatically slows down the diffusion of polymer segments in return. The core–shell model in which the NP and absorbed polymers diffuse as a single object is validated here at the full atomistic level. These results provide atomistic insights into the unique structure and dynamics in the small attractive NP–polymer interfacial region. We hope these results will be helpful for the understanding of peculiar phenomena in attractive polymer nanocomposites containing small NPs.
 Please wait while we load your content...
Please wait while we load your content...

