
Density functional theory study of superoxide ions as impurities in alkali halides†

Abstract
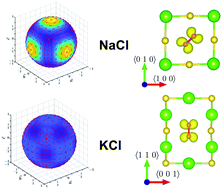
The orientation of diatomic molecular impurities in crystals is a classic problem in physics, whose analysis started in the early 1930s with Pauling's pioneering studies and has extended to the present day. In the present work, we investigate the orientation of a superoxide ion (O2−), which is known to be oriented in the 〈1 1 0〉 direction when replacing a halide ion in alkali halide rock salt lattices. The unpaired electron of the superoxide, whose ground state is degenerate (2Πg), is oriented in the 〈0 0 1〉 direction for sodium halides while it is oriented in the 〈1 ![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0〉 direction for potassium and rubidium halides. We performed density functional theory (DFT) calculations to describe the full adiabatic potential energy surface (APES) of this complex system for the first time with ab initio methods. We are focused on four alkali halide lattices, namely NaCl, NaBr, KCl, and KBr. We show that DFT, at the generalized gradient approximation (GGA) and meta-GGA levels, is able to reproduce all the experimental features for potassium halides. However, for sodium halides, although the DFT predicts the correct unpaired electron orientation, the forecasted APES energy minimum for the molecular orientation is found to be close to the 〈1 1 3/4〉 orientation, in contrast to the experimental 〈1 1 0〉 orientation. The difference in energy between the 〈1 1 3/4〉 and 〈1 1 0〉 orientation is less than 10 meV, which points out the subtleness of the considered problem. In addition to assessing the DFT accuracy and limitations to treat these systems, we also paid special attention to analyze the geometry distortions of the host lattice for the high symmetry orientations of the superoxide ion, i.e., 〈1 0 0〉, 〈1 1 0〉 and 〈1 1 1〉. In the case of the 〈1 1 0〉 molecular orientation, we find a strong dependence on the distance between the alkali ions in the 〈0 0 1〉 direction and the superoxide ion upon the unpaired electron orientation. This fact explains why the orientation of the unpaired electron is different in sodium vs. potassium halides. In the case of the 〈1 0 0〉 and 〈1 1 1〉 molecular orientations, we analyze the Jahn–Teller vibronic coupling to find an unusually large vibronic centrifugal term in the latter.
0〉 direction for potassium and rubidium halides. We performed density functional theory (DFT) calculations to describe the full adiabatic potential energy surface (APES) of this complex system for the first time with ab initio methods. We are focused on four alkali halide lattices, namely NaCl, NaBr, KCl, and KBr. We show that DFT, at the generalized gradient approximation (GGA) and meta-GGA levels, is able to reproduce all the experimental features for potassium halides. However, for sodium halides, although the DFT predicts the correct unpaired electron orientation, the forecasted APES energy minimum for the molecular orientation is found to be close to the 〈1 1 3/4〉 orientation, in contrast to the experimental 〈1 1 0〉 orientation. The difference in energy between the 〈1 1 3/4〉 and 〈1 1 0〉 orientation is less than 10 meV, which points out the subtleness of the considered problem. In addition to assessing the DFT accuracy and limitations to treat these systems, we also paid special attention to analyze the geometry distortions of the host lattice for the high symmetry orientations of the superoxide ion, i.e., 〈1 0 0〉, 〈1 1 0〉 and 〈1 1 1〉. In the case of the 〈1 1 0〉 molecular orientation, we find a strong dependence on the distance between the alkali ions in the 〈0 0 1〉 direction and the superoxide ion upon the unpaired electron orientation. This fact explains why the orientation of the unpaired electron is different in sodium vs. potassium halides. In the case of the 〈1 0 0〉 and 〈1 1 1〉 molecular orientations, we analyze the Jahn–Teller vibronic coupling to find an unusually large vibronic centrifugal term in the latter.
- This article is part of the themed collection: 2020 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

