
Photoelectron spectroscopy and computational investigations of the electronic structures and noncovalent interactions of cyclodextrin-closo-dodecaborate anion complexes χ-CD·B12X122− (χ = α, β, γ; X = H, F)†

Abstract
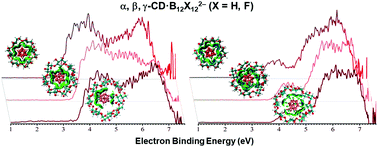
We report a joint negative ion photoelectron spectroscopy (NIPES) and computational study on the electronic structures and noncovalent interactions of a series of cyclodextrin-closo-dodecaborate dianion complexes, χ-CD·B12X122− (χ = α, β, γ; X = H, F). The measured vertical/adiabatic detachment energies (VDEs/ADEs) are 1.15/0.93, 3.55/3.20, 3.90/3.60, and 3.85/3.60 eV for B12H122− and its α-, β-, γ-CD complexes, respectively; while the corresponding values are 1.90/1.70, 4.00/3.60, 4.33/3.95, and 4.30/3.85 eV for the X = F case. These results show that the inclusion of B12X122− into the CD cavities greatly increases the electronic stability of the dianions. The effect of electronic stabilization for β-CD is roughly the same as for γ-CD, both being considerably stronger than that for α-CD. Density functional theory (DFT) based geometry optimization reveals that B12X122− are inserted into CDs increasingly deeper from α-CD to γ-CD. The calculated VDEs and ADEs agree with the experiments well, particularly, reproducing the electron binding energy (EBE) trends. The molecular orbital analyses indicate that the most loosely bound photodetached electrons originate from the guest B12X122− moieties. In addition to a shift of all signals to a larger EBE, significant changes in the signal patterns are observed. At low EBE, this is due to the splitting of highly degenerate B12X122− orbitals, while at high EBE, photodetachment from CD oxygens contributes to the new bands. The guest B12X122− and host CD noncovalent, size-specific interaction based on the independent gradient model (IGM) and energy decomposition analysis (EDA) is dominated by electrostatic interactions. The analysis further unravels unambiguously the existence of dihydrogen bonding and how it affects the total energy that stabilizes the host–guest complexes of CDs·B12H122− compared to the general hydrogen bonding interaction in CDs·B12F122−. This work clearly exhibits strong influences on the electronic structures of dodecaborates upon clustering with CDs, with both size (α-, β-, and γ-) and molecular (X = H or F) specificities, thus providing critical molecular-level information on the cyclodextrin–closo-dodecaborate interactions of interest to medical applications, e.g., boron neutron capture therapy.
 Please wait while we load your content...
Please wait while we load your content...

