
Thermal activation of methane by MgO+: temperature dependent kinetics, reactive molecular dynamics simulations and statistical modeling†
 c
Sebastian
Brickel,
‡d
Oliver T.
Unke,
§d
Meenu
Upadhyay
d
and
Markus
Meuwly
*d
c
Sebastian
Brickel,
‡d
Oliver T.
Unke,
§d
Meenu
Upadhyay
d
and
Markus
Meuwly
*d
Abstract
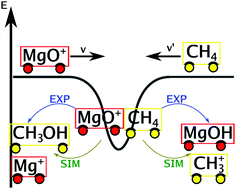
The kinetics of MgO+ + CH4 was studied experimentally using the variable ion source, temperature adjustable selected ion flow tube (VISTA-SIFT) apparatus from 300–600 K and computationally by running and analyzing reactive atomistic simulations. Rate coefficients and product branching fractions were determined as a function of temperature. The reaction proceeded with a rate of k = 5.9 ± 1.5 × 10−10(T/300 K)−0.5±0.2 cm3 s−1. MgOH+ was the dominant product at all temperatures, but Mg+, the co-product of oxygen-atom transfer to form methanol, was observed with a product branching fraction of 0.08 ± 0.03(T/300 K)−0.8±0.7. Reactive molecular dynamics simulations using a reactive force field, as well as a neural network trained on thousands of structures yield rate coefficients about one order of magnitude lower. This underestimation of the rates is traced back to the multireference character of the transition state [MgOCH4]+. Statistical modeling of the temperature-dependent kinetics provides further insight into the reactive potential surface. The rate limiting step was found to be consistent with a four-centered activation of the C–H bond, in agreement with previous calculations. The product branching was modeled as a competition between dissociation of an insertion intermediate directly after the rate-limiting transition state, and traversing a transition state corresponding to a methyl migration leading to a Mg–CH3OH+ complex, though only if this transition state is stabilized significantly relative to the dissociated MgOH+ + CH3 product channel. An alternative, non-statistical mechanism is discussed, whereby a post-transition state bifurcation in the potential surface could allow the reaction to proceed directly from the four-centered TS to the Mg–CH3OH+ complex thereby allowing a more robust competition between the product channels.
- This article is part of the themed collection: Emerging AI Approaches in Physical Chemistry
 Please wait while we load your content...
Please wait while we load your content...

