
Unraveling the structural stability and the electronic structure of ThO2 clusters†

Abstract
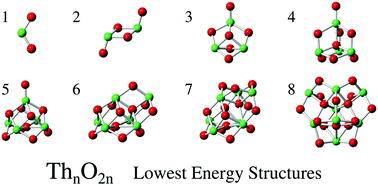
Unraveling the correlations between the geometry, the relative energy and the electronic structure of metal oxide nanostructures is crucial for a better control of their size, shape and properties. In this work, we investigated these correlations for stoichiometric thorium dioxide clusters ranging from ThO2 to Th8O16 using a chemically-driven geometry search algorithm in combination with state-of-the-art first principles calculations. This strategy allows us to homogeneously screen the potential energy surface of actinide oxide clusters for the first time. It is found that the presence of peroxo and superoxo groups tends to increase the total energy of the system by at least 3.5 eV and 7 eV, respectively. For the larger clusters, the presence of terminal oxygen atoms increases the energy by about 0.5 eV. Regarding the electronic structure, it is found that the HOMO–LUMO gap is larger in systems containing only bridging oxygen atoms (∼2–3.5 eV) than for systems containing oxo groups (∼1–3 eV), peroxo groups (∼0–2 eV), and superoxo groups (∼0–1 eV). Furthermore, while the LUMO is always dominated by thorium orbitals, the composition of the HOMO changes in the presence or the absence of oxo, peroxo and/or superoxo groups: in the presence of peroxo groups, it is dominated by thorium orbitals, in all other cases, it is dominated by oxygen orbitals, and is rather localized in the presence of terminal oxo or superoxo groups. These correlations are of great interest for synthesizing clusters with tailored properties, especially for applications in the field of nuclear energy and heterogeneous catalysis.
 Please wait while we load your content...
Please wait while we load your content...

