
Binding affinity and dissociation pathway predictions for a series of USP7 inhibitors with pyrimidinone scaffold by multiple computational methods†

Abstract
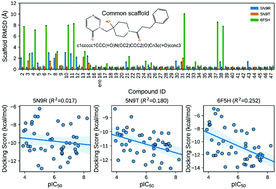
Ubiquitin specific protease 7 (USP7) has attracted increasing attention because of its multifaceted roles in different tumor types. The crystal structures of USP7-inhibitor complexes resolved recently provide reliable models for computational structure-based drug design (SBDD) towards USP7. How to accurately estimate USP7-ligand binding affinity is quite critical to guarantee the reliability of SBDD. In this study, we assessed the reliability of multiple computational methods to the binding affinity prediction for a series of USP7 inhibitors with the pyrimidinone scaffold, including molecular docking scoring, MM/PB(GB)SA, and umbrella sampling (US). It was found that the accuracy of the evaluated computational methods for binding affinity prediction follows the order: US-based method > MM/PB(GB)SA > Glide XP scoring. The calculation results demonstrate that incorporating protein flexibility through induced-fit docking or ensemble docking cannot improve the performance of the Glide scoring based on rigid-receptor docking. For the MM/PB(GB)SA methods, the choice of the protein structure and the calculation procedure has a marked impact on the predictions. More importantly, we discovered for the first time that there are significant differences in the dissociation pathways of strong-binding inhibitors and weak-binding inhibitors of USP7, which may be used as a new criterion to judge whether an inhibitor is a strong binder or not. It is expected that our work can provide valuable guidance on the design and discovery of potent USP7 inhibitors.
- This article is part of the themed collection: 2020 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

