
Structures, stabilities and aromatic properties of endohedrally transition metal doped boron clusters M@B22, M = Sc and Ti: a theoretical study†
 a
Fernando
Buendía,
*b
Alan
Miralrio,
c
Lauro Oliver
Paz-Borbón,
b
Marcela
Beltran,
a
Minh Tho
Nguyen
*d
and
Luis E.
Sansores
a
a
Fernando
Buendía,
*b
Alan
Miralrio,
c
Lauro Oliver
Paz-Borbón,
b
Marcela
Beltran,
a
Minh Tho
Nguyen
*d
and
Luis E.
Sansores
a
Abstract
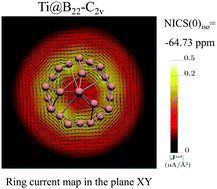
A genetic search algorithm in conjunction with density functional theory calculations was used to determine the lowest-energy minima of the pure B22 cluster and thereby to evaluate the capacity of its isomers to form endohedrally doped cages with two transition metal atoms M (M = Sc and Ti). An important charge transfer from metal atoms M to the boron cage takes place, stabilizing the endohedral compounds, as predicted with the genetic algorithm implemented. High-level coupled-cluster theory CCSD(T) calculations were carried out to confirm that the structures found are the lowest-energy isomers. For a deeper understanding of the doping effects and related charge transfer, the best structural motif of the B22 isomers was also determined when the bare cages are in anionic states, such as B222− and B224−. It was found that B22 has an appropriate size, geometric shape and electronic state to host the chosen metal atoms and, consequently, to form stable endohedrally doped compounds Ti@B22 (C2v, 4-Ti) and Sc@B22 (C2v, 5-Sc). The chemical bonding was analyzed in order to understand the molecular orbitals that these novel systems form. The cage aromaticity was evaluated by means of the nuclear independent chemical shift (NICS(0)iso) indices, the isochemical shielding surface (ICSSzz), the anisotropy of the current induced density (ACID) maps, and the magnetic ring current Gauge-Including Magnetically Induced Current (GIMIC) method, indicating that aromaticity plays a crucial role in the stabilization of endohedrally doped boron clusters. Finally, the thermodynamic stability of the latter, using parameters derived from density functional theory (DFT), was evaluated. Ab initio molecular dynamics (AIMD) simulations were performed to elucidate the stability, at high temperature, of the most stable endohedrally doped boron clusters 4-Ti and 5-Sc.
 Please wait while we load your content...
Please wait while we load your content...

