
Predicting reactive sites with quantum chemical topology: carbonyl additions in multicomponent reactions†
 a
Cesar R.
García-Jacas,
b
Pablo
Carpio-Martínez
c
and
Fernando
Cortés-Guzmán
*a
a
Cesar R.
García-Jacas,
b
Pablo
Carpio-Martínez
c
and
Fernando
Cortés-Guzmán
*a
Abstract
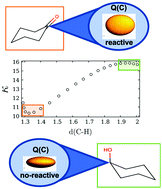
Quantum Chemical Topology (QCT) is a well established structural theoretical approach, but the development of its reactivity component is still a challenge. The hypothesis of this work is that the reactivity of an atom within a molecule is a function of its electronic population, its delocalization in the rest of the molecule, and the way it polarizes within an atomic domain. In this paper, we present a topological reactivity predictor for cabonyl additions, κ. It is a measure of the polarization of the electron density with the carbonyl functional group. κ is a model obtained from a QSAR procedure, using quantum-topological atomic descriptors and reported hydration equilibrium constants of carbonyl compounds. To validate the predictive capability of κ, we applied it to organic reactions, including a multicomponent reaction. κ was the only property that predicts the reactivity in each reaction step. The shape of κ can be interpreted as the change between two electrophilic states of a functional group, reactive and non-reactive.
- This article is part of the themed collection: Celebrating recent chemical science in Mexico
 Please wait while we load your content...
Please wait while we load your content...

