
The usefulness of molecular-dynamics simulations in clarifying the activation enthalpy of oxygen-vacancy migration in the perovskite oxide BaTiO3
Stephen C.
Parker  b
and
Roger A.
De Souza
*a
b
and
Roger A.
De Souza
*a
b
and
Roger A.
De Souza
*a
Abstract
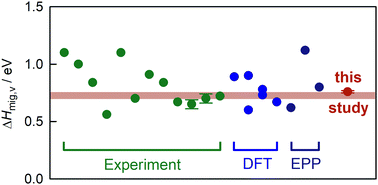
We employed molecular-dynamics simulations with interatomic pair-potentials to examine oxygen-vacancy diffusion in the cubic phase of perovskite BaTiO3 as a function of temperature. By comparing the absolute rate of vacancy diffusion as well as its temperature dependence with experimental data, we are able to narrow down the activation enthalpy of migration to 0.70–0.76 eV.
 Please wait while we load your content...
Please wait while we load your content...

