
Aqueous solvation of the chloride ion revisited with density functional theory: impact of correlation and exchange approximations†

Abstract
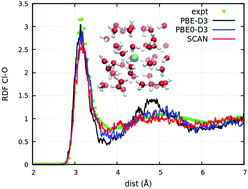
The specificity of aqueous halide solvation is fundamental to a wide range of bulk and interfacial phenomena spanning from biology to materials science. Halide polarizability is thought to drive the ion specificity, and if so, it is essential to have an accurate description of the electronic properties of halide ions in water. To this end, the solvation of the chloride anion, Cl− has been reinvestigated with state-of-the-art density functional theory. Specifically, the PBE-D3, PBE0-D3, and SCAN functionals have been employed to probe the impact of correlation and exchange approximations. Anticipating the findings, adding exact exchange improves the electronic structure, but simultaneously significantly reduces the Cl− polarizability, resulting in an over-structured Cl–O radial distribution function (RDF) and longer water H-bond lifetimes to Cl−. SCAN does not yield as much improvement in the energetics of Cl− relative to bulk water, but does result in a smaller reduction of the polarizability and thus a less structured Cl–O RDF, which agrees better with experiment. Special consideration is therefore warranted in assessing the impact of exchange on the energy, charge density, and the charge density response when designing and testing hybrid functionals for aqueous halide solvation.
- This article is part of the themed collection: Frontiers in Molecular Simulation of Solvated Ions, Molecules and Interfaces
 Please wait while we load your content...
Please wait while we load your content...

