
Is it really possible to control aromaticity of benzene with light?
 *a
and
J. C.
Tremblay
b
*a
and
J. C.
Tremblay
b
Abstract
Recent theoretical investigations claim that tailored laser pulses may selectively steer benzene's aromatic ground state to localized non-aromatic excited states. For instance, it has been shown that electronic wavepackets, involving the two lowest electronic eigenstates, exhibit subfemtosecond charge oscillation between equivalent Kekulé resonance structures. In this contribution, we show that such dynamical electron-localization in the molecule-fixed frame contravenes the principle of the indistinguishability of identical particles. This breach stems from a total omission of the nuclear degrees of freedom, giving rise to nonsymmetric electronic wavepackets under nuclear permutations. Enforcement of the latter leads to entanglement between the electronic and nuclear states. To obey quantum statistics, the entangled molecular states should involve compensating nuclear-permutation symmetries. This in turn engenders complete quenching of dynamical electron-localization in the molecule-fixed frame. Indeed, for the (six-fold) equilibrium geometry of benzene, group-theoretic analysis reveals that any electronic wavepacket exhibits a (D6h) totally symmetric electronic density, at all times. Thus, our results clearly show that the six carbon atoms, and the six C–C bonds, always have equal Mulliken charges, and equal bond orders, respectively. However, electronic wavepackets may display dynamical localization of the electronic density in the space-fixed frame, whenever they involve both even and odd space-inversion (parity) or permutation-inversion symmetry. Dynamical spatial-localization can be probed experimentally in the laboratory frame, but it should not be deemed equivalent to charge oscillation between benzene's identical electronic substructures, such as Kekulé resonance structures.
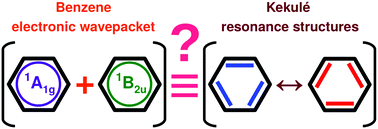
 Please wait while we load your content...
Please wait while we load your content...

