
Hydration in aqueous osmolyte solutions: the case of TMAO and urea

Abstract
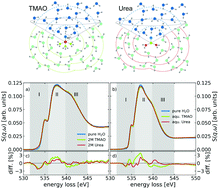
The hydration and hydrogen-bond topology of small water solvated molecules such as the naturally occurring organic osmolytes trimethylamine N-oxide (TMAO) and urea are under intense investigation. We aim at furthering the understanding of this complex hydration by combining experimental oxygen K-edge excitation spectra with results from spectra calculated via the Bethe–Salpeter equation based on structures obtained from ab initio molecular dynamics simulations. Comparison of experimental and calculated spectra allows us to extract detailed information about the immediate surrounding of the solute molecules in the solvated state. We quantify and localize the influence of the solute on the hydrogen bond network of the water solvent and find spectroscopic fingerprints of a clear directional asymmetry around TMAO with strong and local kosmotropic influence around TMAO's NO head group and slight chaotropic influence around the hydrophobic methyl groups. The influence of urea on the local water network is qualitatively similar to that of TMAO but weaker in magnitude. The strongest influence of both molecules on the shape of the oxygen K-edge spectra is found in the first hydration shells.
 Please wait while we load your content...
Please wait while we load your content...

