
Ab initio modelling of local interfaces in doped organic semiconductors†

Abstract
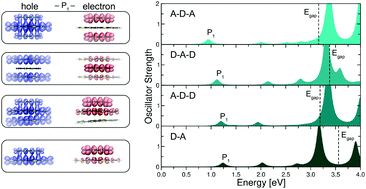
Doping in organic semiconductors remains a debated issue from both an experimental and ab initio perspective. Due to the complexity of these systems, which exhibit a low degree of crystallinity and high level of disorder, modelling doped organic semiconductors from first-principles calculations is not a trivial task, as their electronic and optical properties are sensitive to the choice of initial geometries. A crucial aspect to take into account, in view of rationalizing the electronic structure of these materials through ab initio calculations, is the role of local donor/acceptor interfaces. We address this problem in the framework of state-of-the-art density-functional theory and many-body perturbation theory, investigating the structural, electronic, and optical properties of quaterthiophene and sexithiophene oligomers doped by 2,3,5,6-tetrafluoro-7,7,8,8-tetracyano-quinodimethane (F4TCNQ). We consider different model structures ranging from isolated dimers and trimers, to periodic stacks. Our results demonstrate that the choice of the initial geometry critically impacts the resulting electronic structure and the degree of charge transfer in the materials, depending on the amount and on the nature of the local interfaces between donor and acceptor species. The optical spectra appear less sensitive to these parameters at least from a first glance, although a quantitative analysis of the excitations reveals that their Frenkel or charge-transfer character is affected by the characteristics of the donor/acceptor interfaces as well as by the donor length. Our findings represent an important step forward towards an insightful first-principles description of the microscopic properties of doped organic semiconductors complementary to experiments.
 Please wait while we load your content...
Please wait while we load your content...

