
Revisiting the Volmer–Heyrovský mechanism of hydrogen evolution on a nitrogen doped carbon nanotube: constrained molecular dynamics versus the nudged elastic band method†
 a
Kari
Laasonen
*a
a
Kari
Laasonen
*a
Abstract
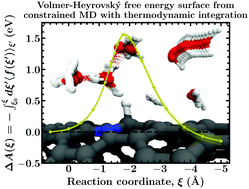
Density functional theory (DFT) based computational electrochemistry has the potential to serve as a tool with predictive power in the rational development and screening of electrocatalysts for renewable energy technologies. It is, however, of paramount importance that simulations are conducted rigorously at a level of theory that is sufficiently accurate in order to obtain physicochemically sensible results. Herein, we present a comparative study of the performance of the static climbing image nudged elastic band method (CI-NEB) vs. DFT based constrained molecular dynamics simulations with thermodynamic integration in estimating activation and reaction (free) energies of the Volmer–Heyrovský mechanism on a nitrogen doped carbon nanotube. Due to cancellation of errors within the CI-NEB calculations, static and dynamic activation barriers are observed to be surprisingly similar, while a substantial decrease in reaction energies is seen upon incorporation of solvent dynamics. This finding is attributed to two competing effects; (1) solvent reorganization that stabilizes the transition and, in particular, the product states with respect to the reactant state and (2) destabilizing entropic contributions due to solvent fluctuations. Our results highlight the importance of explicitly sampling the interfacial solvent dynamics when studying hydrogen evolution at solid–liquid interfaces.
- This article is part of the themed collections: 2020 PCCP HOT Articles and Frontiers in Molecular Simulation of Solvated Ions, Molecules and Interfaces
 Please wait while we load your content...
Please wait while we load your content...

