
Effect of anion reorientation on proton mobility in the solid acids family CsHyXO4 (X = S, P, Se, y = 1, 2) from ab initio molecular dynamics simulations†
 a
and
Daniel
Sebastiani
*a
a
and
Daniel
Sebastiani
*a
Abstract
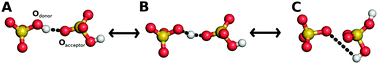
The high temperature phases of the solid acids CsHSeO4, CsHSO4 and CsH2PO4 show extraordinary high proton conductivities, while the low temperature phases do not conduct protons at all. We systematically investigate proton dynamics in the low and high temperature phases of these compounds by means of ab initio molecular dynamics simulations in order to develop a general picture of the proton transfer mechanism. For all of these compounds, proton conduction follows a Grotthuss mechanism via a combined proton transfer and subsequent structural reorientation of the environment. We demonstrate that the drastically reduced conductivity of the low temperature phases is caused by a highly ordered, rigid hydrogen bond network, while efficient long range proton transfer in the high temperature phases is enabled by the interplay of high proton transfer rates and frequent anion reorientation. Furthermore, we present a simple descriptor for the quantitative prediction of the diffusion coefficient within the solid acids family. As a side result, we show that the rate of the most elementary proton hopping reaction depends on the heavy-atom configuration of the nearest atoms in a ubiquitous manner, and is in turn almost independent from the global nature of the compound, i.e. whether it is organic or inorganic, ordered or disordered.
- This article is part of the themed collection: Frontiers in Molecular Simulation of Solvated Ions, Molecules and Interfaces
 Please wait while we load your content...
Please wait while we load your content...

