
Improving the performance of the MM/PBSA and MM/GBSA methods in recognizing the native structure of the Bcl-2 family using the interaction entropy method†
 a
Lili
Duan
*a
a
Lili
Duan
*a
Abstract
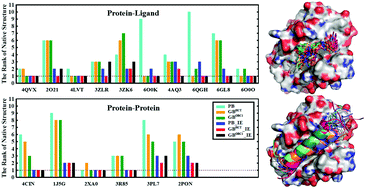
In the research and development of new drugs, theoretical and computational studies play an increasingly important role in discriminating native and decoy structures by their binding free energies. Predicting the binding free energy using the molecular mechanics/Poisson–Boltzmann (Generalized Born) surface area (MM/PB(GB)SA) methods to identify the native structure as the lowest-energy conformation is more theoretically rigorous than most scoring functions, but the main challenge of this method is the calculation of the entropic contribution. In this study, we add the entropic contribution to the MM/PBSA and two MM/GBSA (GBHCT and GBOBC1) models using the interaction entropy (IE) method. We then systemically evaluate the performance of these methods in recognizing the native structures by predicting the binding affinities of 176 protein–ligand and protein–protein systems of the Bcl-2 family. By calculating a series of statistical metrics, sensitivity, specificity, accuracy, Matthews correlation coefficient, the G-mean, and the receiver operating characteristic (ROC) curve, we find that the ability to discern the native structure from a decoy ensemble is improved significantly by the modification of the binding free energy using the IE method in both protein–ligand and protein–protein systems. Furthermore, the maximum area under the ROC curve (AUC) was 0.97, which was obtained by the GBHCT model combined with the IE method, indicating that this method has the best performance. The largest improvement occurs in the PB method, with a change in the AUC of 0.32. The modification of the energy is more obvious for protein–protein interactions than for protein–ligand interactions. This study indicates the effectiveness of the IE method in successfully recognizing the native structure, which is critical in rational drug design.
 Please wait while we load your content...
Please wait while we load your content...

