
 a
E. A.
Bormotova,
a
A. A.
Medvedev,
a
E. A.
Pazyuk,
a
A. V.
Stolyarov
*a
and
A.
Zaitsevskii
ab
a
E. A.
Bormotova,
a
A. A.
Medvedev,
a
E. A.
Pazyuk,
a
A. V.
Stolyarov
*a
and
A.
Zaitsevskii
ab
Abstract
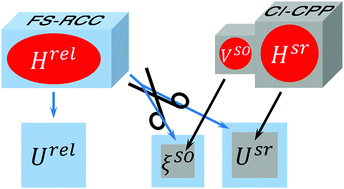
The spin–orbit (SO) interactions in low-lying electronic states of the LiM (M = Na, K, Rb, Cs) molecular series are studied through ab initio calculations of potential energy curves and SO coupling matrix elements as functions of the interatomic distance, R. Two different approaches are employed: (a) the Fock-space relativistic coupled-cluster calculations (FS-RCC) which directly yield full relativistic energies, Urel(R); the SO coupling functions, ξso(R), are extracted a posteriori through projecting scalar-relativistic wave functions onto the subspaces spanned by their full-relativistic counterparts; (b) the evaluation of the scalar-relativistic electronic energies, Usr(R), and relevant ξso(R) functions using the configuration interaction method with core-valence correlation accounted for using core polarization potentials (CI-CPP). The SO-free potentials and SO coupling functions obtained within the framework of both approaches are in good agreement with each other and their prior theoretical and empirical counterparts.
 Please wait while we load your content...
Please wait while we load your content...

