
Exciton transfer free energy from Car–Parrinello molecular dynamics†
 a
and
Nikos L.
Doltsinis
*a
a
and
Nikos L.
Doltsinis
*a
Abstract
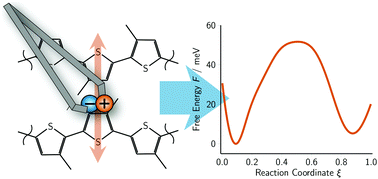
A computational approach is presented which allows the calculation of free energies profiles for exciton transfer processes within the framework of ab initio molecular dynamics (AIMD) simulations, sampling both the electronic and the nuclear degrees of freedom. To achieve this, restraining potentials are imposed on the centres of maximally localized Wannier orbitals. The resulting quantum-mechanical orbital forces are derived analytically and implemented in an AIMD program. In analogy to classical umbrella sampling techniques, these restraints are used to control an exciton transfer by incrementally moving the Wannier centres corresponding to the electron–hole pair along a suitable reaction coordinate. The new method is applied to study exciton transfer between two stacked penta(3-methylthiophene) molecules as a function of intermolecular distance. From the resulting free energy profiles, exciton transfer rates and diffusion constants are estimated, which prove to be in line with experimental results.
- This article is part of the themed collections: 2020 PCCP HOT Articles and Frontiers in Molecular Simulation of Solvated Ions, Molecules and Interfaces
 Please wait while we load your content...
Please wait while we load your content...

