
Vapour–liquid phase equilibria and interfacial properties of fatty acid methyl esters from molecular dynamics simulations

Abstract
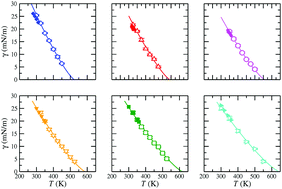
We have determined the phase equilibria and interfacial properties of a methyl ester homologous series (from methyl acetate to methyl heptanoate) using direct simulations of the vapour–liquid interfaces. The methyl esters are modelled using the united atom approach in combination with transferable parameters for phase equilibria (TraPPE) force fields for alkanes, alkenes, carbon dioxide, ethers, and carboxylic acids in a transferable way. This allows us to take into account explicitly both dispersive and coulombic interactions, as well as the repulsive Pauli-exclusion interactions. Simulations are performed in the NVT or canonical ensemble using molecular dynamics. Vapour–liquid surface tension is determined using the virial route, i.e., evaluating the normal and tangential components of the pressure tensor along the simulation box. We have also calculated density profiles, coexistence densities, vapour pressures, surface entropies and enthalpies, and interfacial thickness as functions of temperature, as well as the normal boiling temperatures and the critical temperatures, densities, and pressures for each member of the series. Special attention is paid to the comparison between experimental data taken from the literature and our results obtained using molecular dynamics simulations. We also analyze the effect of increasing the molecular weight of the methyl esters (at fixed temperature) on all the properties considered, with special emphasis on phase equilibria envelopes and surface tension. The TraPPE force fields transferred from other molecules and chemical families are able to predict very accurately the experimental vapour–liquid phase envelopes of methyl esters. We also compare the results obtained from simulations of the surface tension, with experimental data taken from the literature. To our knowledge, this is the first time that vapour–liquid phase equilibria and interfacial properties, and particularly surface tension, of this methyl ester homologous series are obtained using computer simulation.
- This article is part of the themed collection: 2020 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

