
First-principles comparative study of perfect and defective CsPbX3 (X = Br, I) crystals†

Abstract
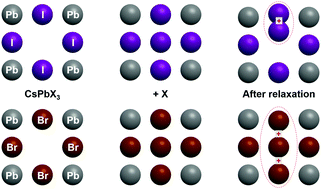
First principles Density Functional Theory (DFT) hybrid functional PBESOL0 calculations of the atomic and electronic structure of perfect CsPbI3, CsPbBr3 and CsPbCl3 crystals, as well as defective CsPbI3 and CsPbBr3 crystals are performed and discussed. For the perfect structure, decomposition energy into binary compounds (CsX and PbX2) is calculated, and a stability trend of the form CsPbBr3 > CsPbI3 > CsPbCl3 is found. In addition, calculations of the temperature-dependent heat capacity are performed and shown to be in good agreement with experimental data. As far as the defect structure is considered, it is shown that interstitial halide atoms in CsPbBr3 do not tend to form di-halide dumbbells Br2− while such dimers are energetically favoured in CsPbI3, analogous to the well-known H-centers in alkali halides. In the case of CsPbBr3, a loose trimer configuration (Br32−) seems to be energetically preferred. The effects of crystalline symmetry and covalency are discussed, alongside the role of defects in recombination processes.
 Please wait while we load your content...
Please wait while we load your content...

