
Charge fluctuations from molecular simulations in the constant-potential ensemble
 a
David T.
Limmer,
bcde
Alessandro
Coretti,
fg
Sara
Bonella,
g
Mathieu
Salanne
ai
and
Benjamin
Rotenberg
*ai
a
David T.
Limmer,
bcde
Alessandro
Coretti,
fg
Sara
Bonella,
g
Mathieu
Salanne
ai
and
Benjamin
Rotenberg
*ai
Abstract
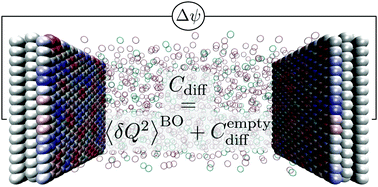
We revisit the statistical mechanics of charge fluctuations in capacitors. In constant-potential classical molecular simulations, the atomic charges of electrode atoms are treated as additional degrees of freedom which evolve in time so as to satisfy the constraint of fixed electrostatic potential for each configuration of the electrolyte. The present work clarifies the role of the overall electroneutrality constraint, as well as the link between the averages computed within the Born–Oppenheimer approximation and that of the full constant-potential ensemble. This allows us in particular to derive a complete fluctuation–dissipation relation for the differential capacitance, that includes a contribution from the charge fluctuations (around the charges satisfying the constant-potential and electroneutrality constraints) also present in the absence of an electrolyte. We provide a simple expression for this contribution from the elements of the inverse of the matrix defining the quadratic form of the fluctuating charges in the energy. We then illustrate numerically the validity of our results, and recover the expected continuum result for an empty capacitor with structureless electrodes at large inter-electrode distances. By considering a variety of liquids between graphite electrodes, we confirm that this contribution to the total differential capacitance is small compared to that induced by the thermal fluctuations of the electrolyte.
- This article is part of the themed collections: 2020 PCCP HOT Articles and Frontiers in Molecular Simulation of Solvated Ions, Molecules and Interfaces
 Please wait while we load your content...
Please wait while we load your content...

