
Hindered rotational barriers in conjugated donor–acceptor substituted systems: calculations vs. experiments†

Abstract
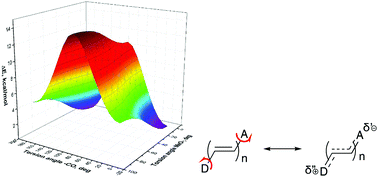
Quantum mechanical calculations of barriers to rotation within push–pull π-conjugated molecules involving strong electron donors (D) and acceptors (A) using the generally accepted approach fail to reproduce the experimental barriers determined by temperature-dependent NMR spectra. On the examples of seven derivatives of this type with substituents of varying electron donating and accepting strength, we find that determination of one of the rotational barriers, for instance, that of the acceptor substituent, requires not only the energy calculation of the respective transition state of this substituent, but also the transition state of the donor and the transition state involving both donor and acceptor substituents. Calculations of the rotation barriers using B3LYP and APFD functionals considering three transition states produce the results with mean absolute deviations from 10 experimental barriers of 0.28–0.19 kcal mol−1 depending on the basis set.
 Please wait while we load your content...
Please wait while we load your content...

