
Quantifying the hydration structure of sodium and potassium ions: taking additional steps on Jacob's Ladder
 *ab
Gregory K.
Schenter,
a
Mirza
Galib,
a
Marcel D.
Baer,
a
X. S.
Zhao
*b
and
Christopher J.
Mundy
*ah
*ab
Gregory K.
Schenter,
a
Mirza
Galib,
a
Marcel D.
Baer,
a
X. S.
Zhao
*b
and
Christopher J.
Mundy
*ah
Abstract
The ability to reproduce the experimental structure of water around the sodium and potassium ions is a key test of the quality of interaction potentials due to the central importance of these ions in a wide range of important phenomena. Here, we simulate the Na+ and K+ ions in bulk water using three density functional theory functionals: (1) the generalized gradient approximation (GGA) based dispersion corrected revised Perdew, Burke, and Ernzerhof functional (revPBE-D3) (2) the recently developed strongly constrained and appropriately normed (SCAN) functional (3) the random phase approximation (RPA) functional for potassium. We compare with experimental X-ray diffraction (XRD) and X-ray absorption fine structure (EXAFS) measurements to demonstrate that SCAN accurately reproduces key structural details of the hydration structure around the sodium and potassium cations, whereas revPBE-D3 fails to do so. However, we show that SCAN provides a worse description of pure water in comparison with revPBE-D3. RPA also shows an improvement for K+, but slow convergence prevents rigorous comparison. Finally, we analyse cluster energetics to show SCAN and RPA have smaller fluctuations of the mean error of ion–water cluster binding energies compared with revPBE-D3.
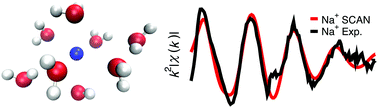
- This article is part of the themed collection: Frontiers in Molecular Simulation of Solvated Ions, Molecules and Interfaces
 Please wait while we load your content...
Please wait while we load your content...

