
Calculation of vibrationally resolved absorption and fluorescence spectra of the rylenes†
 a
and
Dage
Sundholm
*b
a
and
Dage
Sundholm
*b
Abstract
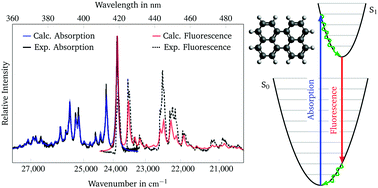
A generating function method was used to simulate the vibrationally resolved absorption and emission spectra of perylene, terrylene and quaterrylene. This method operates on the basis of adiabatic excitation energies and electronic ground and excited state vibrational frequencies. These parameters were calculated using density functional theory with the PBE0 functional for perylene and terrylene and with the BH-LYP functional for quaterrylene. The vertical excitation energies of the lower excited states were calculated using functionals with differing amounts of Hartree–Fock exchange. The optimal functional for each molecule was chosen by comparing these energies to literature excitation energies. Using this technique the calculated absorption spectra and the calculated emission spectrum of perylene were found to be in excellent agreement with the literature experimental spectra after introducing a shift and a scaling factor. The most prominent bands of the absorption spectra were assigned to their respective vibronic transitions.
 Please wait while we load your content...
Please wait while we load your content...

