
The N(4S) + O2(X3Σ −g) ↔ O(3P) + NO(X2Π) reaction: thermal and vibrational relaxation rates for the 2A′, 4A′ and 2A′′ states†

Abstract
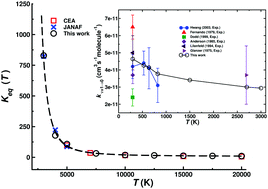
The kinetics and vibrational relaxation of the N(4S) + O2(X3Σ−g) ↔ O(3P) + NO(X2Π) reaction is investigated over a wide temperature range based on quasiclassical trajectory simulations on 3-dimensional potential energy surfaces (PESs) for the lowest three electronic states. Reference energies at the multi reference configuration interaction level are represented as a reproducing kernel and the topology of the PESs is rationalized by analyzing the CASSCF wavefunction of the relevant states. The forward rate matches one measurement at 1575 K and is somewhat lower than the high-temperature measurement at 2880 K whereas for the reverse rate the computations are in good agreement for temperatures between 3000 and 4100 K. The temperature-dependent equilibrium rates are consistent with results from JANAF and CEA results. Vibrational relaxation rates for O + NO(ν = 1) → O + NO(ν = 0) are consistent with a wide range of experiments. This process is dominated by the dynamics on the 2A′ and 4A′ surfaces which both contribute similarly up to temperatures T ∼ 3000 K, and it is found that vibrationally relaxing and non-relaxing trajectories probe different parts of the potential energy surface. The total cross section depending on the final vibrational state monotonically decreases which is consistent with early experiments and previous simulations but at variance with other recent experiments which reported an oscillatory cross section.
 Please wait while we load your content...
Please wait while we load your content...

