
Model systems for screening and investigation of lithium metal electrode chemistry and dendrite formation†

Abstract
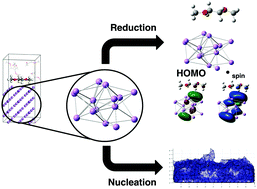
The use of lithium metal as an electrode for electrochemical energy storage will provide a significant impact on practical energy storage technology. Unfortunately, the use of lithium metal is plagued with challenging chemical problems. Specifically, the formation of a solid electrolyte interphase layer and the nucleation and growth of lithium dendrites: both must be addressed and controlled in order to achieve a practically useable pure lithium metal electrode. Currently sophisticated experimental techniques and computationally expensive simulations are being used to probe these problems but these methods are arduous and time consuming which delays timely evaluation and insight into the rapidly changing field of advanced energy storage. Here, we report the use of DFT simulations of lithium nanoclusters to investigate and explore lithium metal chemistry with inexpensive computational methods to gain greater insight into electrochemical reductions and the nucleation and growth of dendrites. DME, LiTFSI, and LiFSI reduction energetics and structures with electrode effects from lithium metal are reported providing better physical description of the absolute reduction potential characterization. The electronic structure of the lithium nanoclusters were used to investigate the nucleation and growth of lithium dendrites from an ab initio perspective. The results demonstrate that kinetic processes have more control over non uniform growth than thermodynamic processes. Based on this information, a non ab initio model was created in Matlab that shows the initial stages of dendrite nucleation considering approximately 2000 atoms.
- This article is part of the themed collection: 2019 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

