
Mechanisms and kinetics of the low-temperature oxidation of 2-methylfuran: insight from DFT calculations and kinetic simulations†
 *a
*a
Abstract
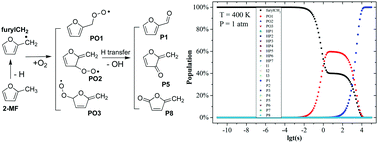
The low-temperature oxidation (LTO) mechanisms of the 2-methylfuran (2-MF) biofuel and the corresponding thermodynamic and kinetic properties have been explored by density functional theory (DFT) and composite G4 methodologies as well as kinetic simulations. The O2 addition to the main furylCH2 radical from the methyl dehydrogenation in 2-MF forms three peroxide radicals PO1, PO2, and PO3 with the energy barriers of 15.1, 19.3, and 20.6 kcal mol−1 and the reaction ΔG of −8.2, 5.7, and −0.1 kcal mol−1 (298 K and 1 atm), respectively. Through hydrogen transfer followed by dehydroxylation, these nascent products evolve into stable aldehydes and cyclic ketones, which may further decompose into smaller species under the action of OH. Calculations and simulations show that the product P1 from the dehydroxylation of PO1 has a dominant population (higher than 96%) among the final products, although the temperature and pressure may influence the species profiles and rate constants to some extent. Based on the G4-calibrated thermodynamic parameters, the temperature and pressure dependence of the rate constants and the two- and three-parameter Arrhenius coefficients for all reactions considered here have been determined by using the transition state theory (TST) and Rice–Ramsperger–Kassel–Marcus (RRKM) methods. The present results provide a comprehensive understanding of the mechanisms and kinetics of the LTO process of the 2-MF biofuel.
 Please wait while we load your content...
Please wait while we load your content...

