
Molecular dynamics simulation of imidazolium CnMIM-BF4 ionic liquids using a coarse grained force-field

Abstract
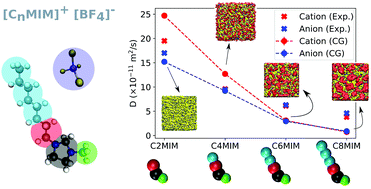
Ionic liquids feature thermophysical properties that are of interest in solvents, energy storage materials and tunable lubrication applications. Here we use new Coarse Grained (CG) models to investigate the structure, dynamics and interfacial properties of the [C2–8MIM][BF4] family of ionic liquids (ILs). The simulated equation of state and diffusion coefficients are in good agreement with experimental data and with all-atom force-fields. We quantify the nano-structure and liquid–vapour interfacial properties of the ILs as a function of the size of the imidazolium cation. The computational efficiency of the CG models enables the simulation of very long time scales (100's of nanoseconds), which are needed to resolve the dynamic and interfacial properties of ILs containing cations with long aliphatic chains. For [C>4MIM] [BF4] the break in symmetry associated to the liquid–vapour interface induces nanostructuring of polar and non-polar domains in the direction perpendicular to the interface plane, with the inhomogeneous regions penetrating deep inside the bulk liquid, typically 5 nm for C8MIM cations.
 Please wait while we load your content...
Please wait while we load your content...

