
Understanding the electrochemistry of armchair graphene nanoribbons containing nitrogen and oxygen functional groups: DFT calculations†
 *a
Juan L.
Fajardo-Díaz,
a
Alejandro J.
Cortés-López,
a
Cristina L.
Rodríguez-Corvera,
a
Luis E.
Jiménez-Ramírez
a
and
Emilio
Muñoz-Sandoval
a
*a
Juan L.
Fajardo-Díaz,
a
Alejandro J.
Cortés-López,
a
Cristina L.
Rodríguez-Corvera,
a
Luis E.
Jiménez-Ramírez
a
and
Emilio
Muñoz-Sandoval
a
Abstract
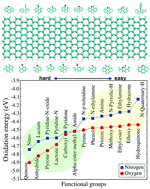
The surface and edge chemistry are vital points to assess a specific application of graphene and other carbon nanomaterials. Based on first-principles density functional theory, we investigate twenty-four chemical functional groups containing oxygen and nitrogen atoms anchored to the edges of armchair graphene nanoribbons (AGNRs). Results for the band structures, formation energy, band gaps, electronic charge deficit, oxidation energy, reduction energy, and global hydrophilicity index are analyzed. Among the oxygen functional groups, carbonyl, anhydride, quinone, lactone, phenol, ethyl-ester, carboxyl, α-ester-methyl, and methoxy act as electron-withdrawing groups and, conversely, pyrane, pyrone, and ethoxy act as electron-donating groups. In the case of nitrogen-functional groups, amine, N-p-toluidine, ethylamine, pyridine-N-oxide, pyridone, lactam, and pyridinium transfer electrons to the AGNRs. Nitro, amide, and N-ethylamine act as electron-withdrawing groups. The carbonyl and pyridinium group-AGNRs show metallic behavior. The formation energy calculations revealed that AGNRs with pyridinium, amine, pyrane, carbonyl, and phenol are the most stable structures. In terms of the global hydrophilicity index, the quinone and N-ethylamine groups showed the most significant values, suggesting that they are highly efficient in accepting electrons from other chemical species. The oxidation and reduction energies as a function of the ribbon's width are discussed for AGNRs with quinone, hydroquinone, nitro, and nitro + 2H. Besides, we discuss the effect of nitrogen-doping in AGNRs on the oxidation and reduction energies for the quinone and hydroquinone functional groups.
- This article is part of the themed collection: Celebrating recent chemical science in Mexico
 Please wait while we load your content...
Please wait while we load your content...

