
From the perspectives of DFT calculations, thermodynamic modeling, and kinetic Monte Carlo simulations: the interaction between hydrogen and Sc2C monolayers†

Abstract
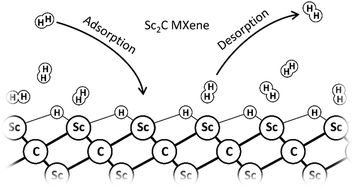
In this study, we have examined the adsorption properties of hydrogen on pristine Sc2C monolayers by DFT calculations. Based on these calculations, we have proposed a thermodynamic model to estimate the hydrogen storage capability within the typical ranges for the operating temperature and pressure. Our thermodynamic modeling has shown that the maximum uptake of usable hydrogen could reach up to 7.2 wt% under cryogenic conditions. When calculating the usable hydrogen uptake, we have taken into consideration that, under realistic operating conditions, not all hydrogen adsorbed on pristine Sc2C can be desorbed from the surface, as some surface–adsorbate interactions are too strong. On the other hand, the interaction between the usable hydrogen and Sc2C appears to be too weak to reach the targets for the year 2025 set by the US Department of Energy (5.5 wt% at operating temperatures between 233 K and 358 K and delivery pressures of up to 12 bar). According to the modeling results, one needs to decrease the temperature to 120 K to reach 5.5 wt% hydrogen uptake at 12 bar. The results obtained with the thermodynamic model have been confirmed with a kinetic Monte Carlo simulation, which has also been used to estimate the time scale of the hydrogen adsorption and desorption processes. In addition, we have also evaluated the changes in the electronic structure of the Sc2C monolayer upon adsorbing hydrogen. As the band gap of Sc2C changes significantly upon adsorbing H2, Sc2C may have more potential as a hydrogen detector instead of as a hydrogen storage material.
 Please wait while we load your content...
Please wait while we load your content...

