
Computational design of p-(dimethylamino)benzylidene-derived push–pull polyenes with high first-hyperpolarizabilities†

Abstract
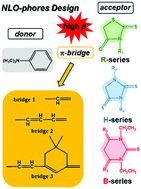
Multiple theoretical investigations on three new series of donor–bridge–acceptor substituted compounds are employed to aid in the design of NLO-phores with high first-hyperpolarizability β. The effect of varying the acceptor (rhodanine, thiohydantoin and thiobarbituric acid derivative-based) and bridge parts of these D–π–A systems was analyzed in terms of geometric and optoelectronic parameters such as bond length alternation, ground state dipole moments, HOMO and LUMO energies, UV-vis absorption spectra, transition dipole moments, and electronic absorption energies. Various functionals with the AUG-cc-pVDZ basis set including B3LYP, PBE38, and ωB97XD, and the Hartree–Fock method were employed to calculate β values, and the solvent effect was also considered by employing the SMD model. The variation of first-hyperpolarizabilities has been explained satisfactorily in terms of the PBE38/AUG-cc-pVDZ level calculated spectroscopic properties in the light of the sum-over-states method and the two-level model. The comprehensive study indicates that the most worthwhile targets for development as NLO-phores are compounds that include a longer π-bridge.
 Please wait while we load your content...
Please wait while we load your content...

