
Recognition of PDL1/L2 by different induced-fit mechanisms of PD1: a comparative study of molecular dynamics simulations†

Abstract
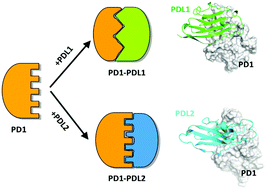
Recent clinical data has shown that some cancers choose to express PDL2 compared to PDL1. Therefore, a detailed and comparative study of the dynamic binding mechanism between PD1/PDL1 and PD1/PDL2 can guide drug design towards PD1. Herein, long time-scale classical molecular dynamics simulation, binding free energy calculation, energy decomposition and homology modeling for PD1/PDL2 were used to shed light on the differences in the binding mechanisms of the PD1/PDL1 and PD1/PDL2 complexes. On one hand, our results reveal a different binding mechanism of PD1 binding to PDL1 and PDL2, which is mainly attributed to the induced-fit from different proteins, that is, the C′D loop of PD1 is essential for PD1/PDL1, while the CD loop of PDL2 is critical for PD1/PDL2. Particularly, the “enclosed” conformation of PDL2 leads to a higher affinity between PD1–PDL2 in comparison to the affinity between PD1–PDL1. For PD1/PDL1, the key residues of N66, Y68, Q75, T76, K78, D85, I126, L128, A132, I134 and E136 are the dominant residues for stabilizing the protein–protein interaction (PPI). For PD1/PDL2, the key residues are mainly concentrated in the FG loop, including N33/Q75/L128/A132/Q133/I134/K135. These findings provide a comprehensive understanding of the distinctive binding kinetics and thermodynamic features, which will contribute meaningfully for the design of peptides and small molecule inhibitors to selectively break the PPI interfaces of PD1/PDL1 and PD1/PDL2.
 Please wait while we load your content...
Please wait while we load your content...

