
DFT study on MgAl-layered double hydroxides with different interlayer anions: structure, anion exchange, host–guest interaction and basic sites†
 *a
*a
Abstract
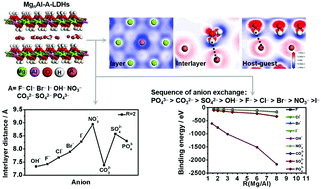
The guest anions play a key role in the construction of layered double hydroxide (LDH)-based host–guest functional materials. In this work, the orientation of the interlayer species, interlayer distances, binding energies, electronic density differences and density of states of the MgnAl-LDHs (n = 1.6, 2.0, 2.6, 3.5, 5.0, and 8.0) with nine different anions (F−, Cl−, Br−, I−, OH−, NO3−, CO32−, SO42−, and PO43−) are calculated by density functional theory (DFT). The results reveal that the LDHs containing the anions with more inclined arrangement, larger size, lower charge and with a larger number of interlayer water molecules show larger interlayer distances. The higher anion charge leads to a larger binding energy for LDHs, and the order of binding energy implies that the sequence of anion exchange is PO43− > CO32− > SO42− > OH− > F− > Cl− > Br− > NO3− > I−. The interactions between interlayer species and the host layer or the interlayer water molecules are mainly derived from the electrostatic interactions. The main components of the valence band maximum (VBM) and conduction band minimum (CBM) of MgAl-LDHs are derived from p orbitals of halogen anions or the O-2p orbitals of other anions, and the Mg-2p orbital, respectively. This illustrates that the most basic sites of MgAl-LDHs are the interlayer anions rather than the hydroxyl group in the layer, while the most acidic sites are Mg in the layers. And LDHs containing anions with higher charge show stronger basicity. The calculation results agree well with the experimental findings. This work provides effective theoretical information for the design and preparation of the anion-controlled functional LDHs or related materials with prospective applications.
 Please wait while we load your content...
Please wait while we load your content...

