
Benchmark ab initio and dynamical characterization of the stationary points of reactive atom + alkane and SN2 potential energy surfaces

Abstract
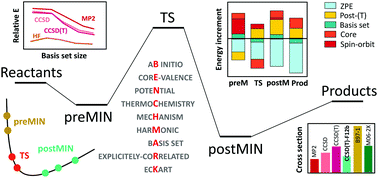
We describe a composite ab initio approach to determine the best technically feasible relative energies of stationary points considering additive contributions of the CCSD(T)/complete-basis-set limit, core and post-CCSD(T) correlation, scalar relativistic and spin–orbit effects, and zero-point energy corrections. The importance and magnitude of the different energy terms are discussed using examples of atom/ion + molecule reactions, such as X + CH4/C2H6 and X− + CH3Y/CH3CH2Cl [X, Y = F, Cl, Br, I, OH, etc.]. We test the performance of various ab initio levels and recommend the modern explicitly-correlated CCSD(T)-F12 methods for potential energy surface (PES) developments. We show that the choice of the level of electronic structure theory may significantly affect the reaction dynamics and the CCSD(T)-F12/double-zeta PESs provide nearly converged cross sections. Trajectory orthogonal projection and an Eckart-transformation-based stationary-point assignment technique are proposed to provide dynamical characterization of the stationary points, thereby revealing front-side complex formation in SN2 reactions and transition probabilities between different stationary-point regions.
- This article is part of the themed collection: PCCP Perspectives
 Please wait while we load your content...
Please wait while we load your content...

