
Properties of the tetravalent actinide series in aqueous phase from a microscopic simulation self-consistent engine†

Abstract
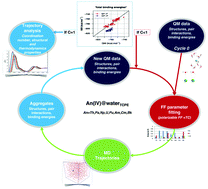
In the context of nuclear fuel recycling and environmental issues, the understanding of the properties of radio-elements with various approaches remains a challenge regarding their dangerousness. Moreover, experimentally, some issues are also of importance; first, it is imperative to work at sufficiently high concentrations to reach the sensitivities of the analytical tools, however this condition often leads to precipitation for some of them; second, stabilizing specific oxidation states of some actinides remains a challenge, thus making it difficult to extract general trends across the actinide series. Complementary to experiments, modeling can be used to unbiasedly probe the actinide's properties in an aquatic environment and offers a predictive tool. We report the first molecular dynamics simulations based on homogeneously built force fields for the whole series of the tetravalent actinides in aqueous phase from ThIV to BkIV and including PuIV. The force fields used to model the interactions among the constituents include polarization and charge donation microscopic effects. They are built from a self-consistent iterative ab initio based engine that can be included in future developments as an element of a potential machine learning procedure devoted to generating accurate force fields. The comparison of our simulated hydrated actinide properties to available experimental data shows the model robustness and the relevance of our parameter assignment engine. Moreover, our simulated structural, dynamical and evolution of the hydration free energy data show that, apart from AmIV and CmIV, the actinide properties change progressively along the series.
 Please wait while we load your content...
Please wait while we load your content...

