
Modeling amino-acid side chain infrared spectra: the case of carboxylic residues†
 ab
Nicola
Tasinato,
a
Vincenzo
Barone,
a
Andrea
Amadei,
c
Isabella
Daidone
*b
ab
Nicola
Tasinato,
a
Vincenzo
Barone,
a
Andrea
Amadei,
c
Isabella
Daidone
*b
Abstract
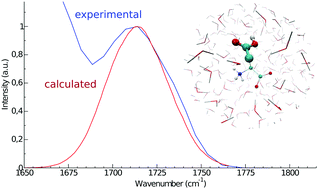
Infrared (IR) spectroscopy is commonly utilized for the investigation of protein structures and protein-mediated processes. While the amide I band provides information on protein secondary structures, amino acid side chains are used as IR probes for the investigation of protein reactions, such as proton pumping in rhodopsins. In this work, we calculate the IR spectra of the solvated aspartic acid, with both zwitterionic and protonated backbones, and of a capped form, i.e. mimicking the aspartic acid residue in proteins, by means of molecular dynamics (MD) simulations and the perturbed matrix method (PMM). This methodology has already proved its good modeling capabilities for the amide I mode and is here extended to the treatment of protein side chains. The computed side chain vibrational signal is in very good agreement with the experimental one, well reproducing both the peak frequency position and the bandwidth. In addition, the MD-PMM approach proposed here is able to reproduce the small frequency shift (5–10 cm−1) experimentally observed between the protonated and zwitterionic forms, showing that such a shift depends on the excitonic coupling between the modes localized on the side chain and on the backbone in the protonated form. The spectrum of the capped form, in which the amide I band is also calculated, agrees well with the corresponding experimental spectrum. The reliable calculation of the vibrational bands of carboxyl-containing side chains provides a useful tool for the interpretation of experimental spectra.
 Please wait while we load your content...
Please wait while we load your content...

