
Electronic structure calculations permit identification of the driving forces behind frequency shifts in transition metal monocarbonyls†
 a
Christianna N.
Lininger,
bc
Alexis T.
Bell,
bc
Teresa
Head-Gordon
abcd
and
Martin
Head-Gordon
*ab
a
Christianna N.
Lininger,
bc
Alexis T.
Bell,
bc
Teresa
Head-Gordon
abcd
and
Martin
Head-Gordon
*ab
Abstract
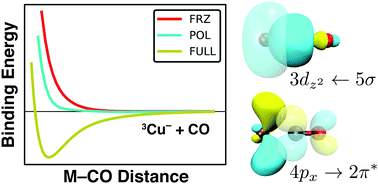
We report the adiabatic energy decomposition analysis (EDA) of density functional theory (DFT) results, shedding light on the physical content of binding energies and carbon monoxide (CO) frequency (υCO) shifts in select first-row transition metal monocarbonyls (MCOs; M = Ti−, V−, Cr−, Co−, Ni−, Cu−, V, Cr, Mn, Ni, Cu, Zn, Cr+, Mn+, Fe+, Cu+, and Zn+). This approach allows for the direct decomposition of υCO, in contrast to previous studies of these systems. Neutral, anionic, and cationic systems are compared, and our results indicate that the relative importance of electrostatic interactions, intramolecular orbital polarization, and charge transfer can vary significantly with the charge and electron configuration of the metal participating in binding. Various anomalous systems are also discussed and incorporated into a general model of MCO binding. Electrostatic interactions and orbital polarization are found to promote blue shifts in υCO, while charge transfer effects encourage υCO red-shifting; previously reported values of υCO are found to be a result of a complex but quantifiable interplay between these physical components. Our computations indicate that CuCO− and ZnCO possess triplet ground states, and also that CrCO− exhibits a non-linear geometry, all in contrast to previous computational results. Advantages and limitations of this model as an approximation to more complicated systems, like those implicated in heterogeneous catalysis, are discussed. We also report benchmark results for MCO geometries, binding energies, and harmonic CO frequencies, and discuss the validity of single-reference wave function and DFT approaches to the study of these transition metal systems.
 Please wait while we load your content...
Please wait while we load your content...

