
A first-principles study on the influences of metal species Al, Zr, Mo and Tc on the mechanical properties of U3Si2
 a
a
Abstract
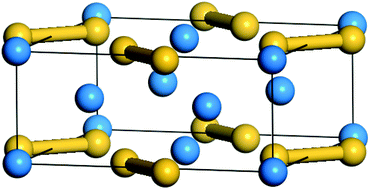
A first-principles approach is employed to study the influences of the metal species Al, Zr, Mo and Tc on the mechanical properties of U3Si2. When the Al, Zr, Mo and Tc atoms diffuse into the vacancy sites, they dissolve into the lattice, as confirmed by the solution energies. It is found that the compounds of U3Si2 with low amounts of Al, Zr, Mo, and Tc in the Si vacancies or Al, Zr, and Mo in the U vacancies can behave in the manner of ductility. However, in the cases where Al, Zr, Mo and Tc occupy the interstitial sites, all the compounds are demonstrated to be brittle. Furthermore, the stress–strain relationship for the U3Si1.9375Mo0.0625 system was calculated, which illustrates the enhanced ductility. The current results indicate that the substitution of Si by carefully selected metal atoms can enhance the performance of U3Si2.
 Please wait while we load your content...
Please wait while we load your content...

