
Structural analysis of metastable pharmaceutical loratadine form II, by 3D electron diffraction and DFT+D energy minimisation†
 *a
Partha P.
Das,
*b
Enrico
Mugnaioli,
c
Iryna
Andrusenko,
c
Jacco
van de Streek,
d
Mauro
Gemmi
c
and
*a
Partha P.
Das,
*b
Enrico
Mugnaioli,
c
Iryna
Andrusenko,
c
Jacco
van de Streek,
d
Mauro
Gemmi
c
and
Abstract
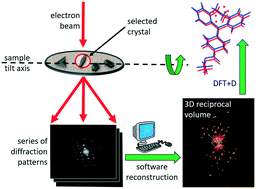
Metastable polymorphs typically display higher solubility than their thermodynamically stable counterparts, whilst having dissimilar mechanical and biopharmaceutical properties. It is unsurprising then, that generic and innovator companies alike pursue isolation and characterisation of these materials. Here we report the determination of the crystal structure of the metastable form II of loratadine using a combination of low-resolution 3D electron diffraction data and density functional theory. Importantly, electron diffraction was able to establish that the crystallites were phase pure i.e. no other polymorphic forms were identified throughout the sample. 3D data collected at room temperature circumvented potential phase changes, conveniently preserving the metastable polymorph during structural elucidation. The limited resolution of the electron diffraction data (>1 Å), combined with the complexity of configurational disorder and possible beam-induced amorphization, meant that the structure could not be obtained by ab initio direct methods. This is a recurrent situation for nanocrystalline pharmaceutical crystals. Instead, two possible starting models arose from simulated annealing based on diffraction data alone. Density functional theory energy minimisation followed, determining the correct model, with an independent validation of the experimental structural solution, comparing favourably to single-crystal X-ray diffraction studies. Our results reveal a promising protocol enabling the exploitation of electron diffraction data with limited resolution obtained from beam sensitive organic materials. The method is widely extendable to a number of pharmaceutical compounds that are not amenable for the growth of large single crystals required by X-ray diffraction, and for which efficient structure determination is required.
- This article is part of the themed collection: The Cambridge Structural Database - A wealth of knowledge gained from a million structures
 Please wait while we load your content...
Please wait while we load your content...

