
Evaluation of top-down mass spectrometry and ion-mobility spectroscopy as a means of mapping protein-binding motifs within heparin chains†
 ‡a
and
Igor A.
Kaltashov
*a
‡a
and
Igor A.
Kaltashov
*a
Abstract
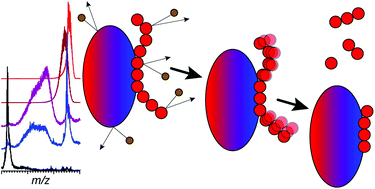
Identifying structural elements within heparin (as well as other glycosaminoglycan) chains that enable their interaction with a specific client protein remains a challenging task due to the high degree of both intra- and inter-chain heterogeneity exhibited by this polysaccharide. The new experimental approach explored in this work is based on the assumption that the heparin chain segments bound to the protein surface will be less prone to collision-induced dissociation (CID) in the gas phase compared to the chain regions that are not involved in binding. Facile removal of the unbound chain segments from the protein/heparin complex should allow the length and the number of sulfate groups within the protein-binding segment of the heparin chain to be determined by measuring the mass of the truncated heparin chain that remains bound to the protein. Conformational integrity of the heparin-binding interface on the protein surface in the course of CID is ensured by monitoring the evolution of collisional cross-section (CCS) of the protein/heparin complexes as a function of collisional energy. A dramatic increase in CCS signals the occurrence of large-scale conformational changes within the protein and identifies the energy threshold, beyond which relevant information on the protein-binding segments of heparin chains is unlikely to be obtained. Testing this approach using a 1 : 1 complex formed by a recombinant form of an acidic fibroblast growth factor (FGF-1) and a synthetic pentasaccharide GlcNS,6S-GlcA-GlcNS,3S,6S-IdoA2S-GlcNS,6S-Me as a model system indicated that a tri-saccharide fragment is the minimal-length FGF-binding segment. Extension of this approach to a decameric heparin chain (dp10) allowed meaningful binding data to be obtained for a 1 : 1 protein/dp10 complex, while the ions representing the higher stoichiometry complex (2 : 1) underwent dissociation via asymmetric charge partitioning without generating truncated heparin chains that remain bound to the protein.
 Please wait while we load your content...
Please wait while we load your content...

