
Molecular mechanisms of thermal instability in hybrid perovskite light absorbers for photovoltaic solar cells†

Abstract
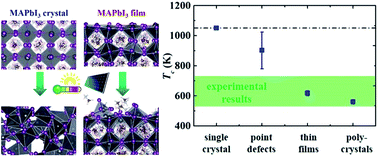
The organic–inorganic hybrid perovskites have been widely explored as key functional components for energy harvesting/conversion applications due to their superior photovoltaic properties. However, material stability issues, such as temperature induced instability of hybrid perovskite crystals during normal device operating conditions, limit their practical application. Here we conduct molecular dynamics simulations to investigate the thermal instability in pristine as well as defective crystals of the prototypical organic–inorganic hybrid perovskite, methylammonium lead iodide (MAPbI3). We show that the accumulation of tilting and splitting of PbI6 octahedra with increasing temperatures initiates the instability in pristine MAPbI3 crystals. Point defects can accelerate the inception of local lattice instability, and the crystals with such defects in the concentration range typically observed in perovskite-based devices undergo an irreversible instability at much lower temperatures. Two-dimensional defects such as grain boundaries in polycrystalline MAPbI3 crystals further decrease their crystal instability temperature to about 550 K, in good agreement with experimental measurements. Finally, we demonstrate that thermal instability in MAPbI3 thin films originates from their free surfaces at much lower temperatures due their increased free energies. We also investigate the structural evolution of the local lattice and show that Born and Lindemann crystal instability criteria coincide in initiating the instability. The key insights obtained from this work can usher a rational design of highly stable hybrid perovskites for their robust and reliable photovoltaic applications.
 Please wait while we load your content...
Please wait while we load your content...

