
Oxygen evolution reaction: a perspective on a decade of atomic scale simulations†

Abstract
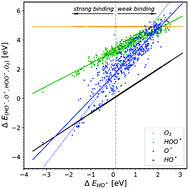
Multiple strategies to overcome the intrinsic limitations of the oxygen evolution reaction (OER) have been proposed by numerous research groups. Despite the substantial efforts, the driving force required for water oxidation is largely making the reaction inefficient. In the present work, we collected published studies involving DFT calculations for the OER, with the purpose to understand why the progress made so far, for lowering the overpotential of the reaction, is relatively small. The data revealed that the universal scaling relationship between HO* and HOO* intermediates is still present and robust, despite the variety in methods and structures used for calculating the binding energies of the intermediates. On the other hand, the data did not show a clear trend line regarding the O* binding. Our analysis suggested that trends in doped semiconducting oxides behave very differently from those in other oxides. This points towards a computational challenge in describing doped oxides in a realistic manner. We propose a way to overcome these computational challenges, which can be applied to simulations corresponding to doped semiconductors in general.
 Please wait while we load your content...
Please wait while we load your content...

