
Total synthesis of biseokeaniamides A–C and late-stage electrochemically-enabled peptide analogue synthesis†

Abstract
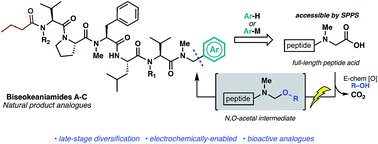
The first total synthesis of cytotoxic cyanobacterial peptide natural products biseokeaniamides A–C is reported employing a robust solid-phase approach to peptide backbone construction followed by coupling of a key thiazole building block. To rapidly access natural product analogues, we have optimized an operationally simple electrochemical oxidative decarboxylation–nucleophilic addition pathway which exploits the reactivity of native C-terminal peptide carboxylates and abrogates the need for building block syntheses. Electrochemically-generated N,O-acetal intermediates are engaged with electron-rich aromatics and organometallic reagents to forge modified amino acids and peptides. The value of this late-stage modification method is highlighted by the expedient and divergent production of bioactive peptide analogues, including compounds which exhibit enhanced cytotoxicity relative to the biseokeaniamide natural products.
- This article is part of the themed collections: Editor’s Choice – Ning Jiao and Celebrating 10 years of Chemical Science
 Please wait while we load your content...
Please wait while we load your content...

