
Stereoretention in styrene heterodimerisation promoted by one-electron oxidants†
 ‡a
and
Robert S.
Paton
*ab
‡a
and
Robert S.
Paton
*ab
Abstract
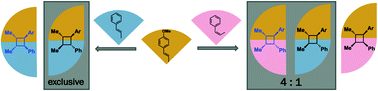
Radical cations generated from the oxidation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C π-bonds are synthetically useful reactive intermediates for C–C and C–X bond formation. Radical cation formation, induced by sub-stoichiometric amounts of external oxidant, are important intermediates in the Woodward–Hoffmann thermally disallowed [2 + 2] cycloaddition of electron-rich alkenes. Using density functional theory (DFT), we report the detailed mechanisms underlying the intermolecular heterodimerisation of anethole and β-methylstyrene to give unsymmetrical, tetra-substituted cyclobutanes. Reactions between trans-alkenes favour the all-trans adduct, resulting from a kinetic preference for anti-addition reinforced by reversibility at ambient temperatures since this is also the thermodynamic product; on the other hand, reactions between a trans-alkene and a cis-alkene favour syn-addition, while exocyclic rotation in the acyclic radical cation intermediate is also possible since C–C forming barriers are higher. Computations are consistent with the experimental observation that hexafluoroisopropanol (HFIP) is a better solvent than acetonitrile, in part due to its ability to stabilise the reduced form of the hypervalent iodine initiator by hydrogen bonding, but also through the stabilisation of radical cationic intermediates along the reaction coordinate.
C π-bonds are synthetically useful reactive intermediates for C–C and C–X bond formation. Radical cation formation, induced by sub-stoichiometric amounts of external oxidant, are important intermediates in the Woodward–Hoffmann thermally disallowed [2 + 2] cycloaddition of electron-rich alkenes. Using density functional theory (DFT), we report the detailed mechanisms underlying the intermolecular heterodimerisation of anethole and β-methylstyrene to give unsymmetrical, tetra-substituted cyclobutanes. Reactions between trans-alkenes favour the all-trans adduct, resulting from a kinetic preference for anti-addition reinforced by reversibility at ambient temperatures since this is also the thermodynamic product; on the other hand, reactions between a trans-alkene and a cis-alkene favour syn-addition, while exocyclic rotation in the acyclic radical cation intermediate is also possible since C–C forming barriers are higher. Computations are consistent with the experimental observation that hexafluoroisopropanol (HFIP) is a better solvent than acetonitrile, in part due to its ability to stabilise the reduced form of the hypervalent iodine initiator by hydrogen bonding, but also through the stabilisation of radical cationic intermediates along the reaction coordinate.
 Please wait while we load your content...
Please wait while we load your content...

