
Prediction of drug metabolites using neural machine translation†

Abstract
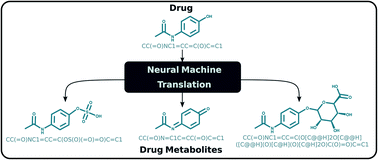
Metabolic processes in the human body can alter the structure of a drug affecting its efficacy and safety. As a result, the investigation of the metabolic fate of a candidate drug is an essential part of drug design studies. Computational approaches have been developed for the prediction of possible drug metabolites in an effort to assist the traditional and resource-demanding experimental route. Current methodologies are based upon metabolic transformation rules, which are tied to specific enzyme families and therefore lack generalization, and additionally may involve manual work from experts limiting scalability. We present a rule-free, end-to-end learning-based method for predicting possible human metabolites of small molecules including drugs. The metabolite prediction task is approached as a sequence translation problem with chemical compounds represented using the SMILES notation. We perform transfer learning on a deep learning transformer model for sequence translation, originally trained on chemical reaction data, to predict the outcome of human metabolic reactions. We further build an ensemble model to account for multiple and diverse metabolites. Extensive evaluation reveals that the proposed method generalizes well to different enzyme families, as it can correctly predict metabolites through phase I and phase II drug metabolism as well as other enzymes. Compared to existing rule-based approaches, our method has equivalent performance on the major enzyme families while it additionally finds metabolites through less common enzymes. Our results indicate that the proposed approach can provide a comprehensive study of drug metabolism that does not restrict to the major enzyme families and does not require the extraction of transformation rules.
- This article is part of the themed collections: Celebrating five years of ChemRxiv and Celebrating 10 years of Chemical Science
 Please wait while we load your content...
Please wait while we load your content...

