

Abstract
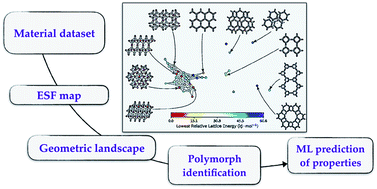
Porous molecular crystals are an emerging class of porous materials formed by crystallisation of molecules with weak intermolecular interactions, which distinguishes them from extended nanoporous materials like metal–organic frameworks (MOFs). To aid discovery of porous molecular crystals for desired applications, energy–structure–function (ESF) maps were developed that combine a priori prediction of both the crystal structure and its functional properties. However, it is a challenge to represent the high-dimensional structural and functional landscapes of an ESF map and to identify energetically favourable and functionally interesting polymorphs among the 1000s to 10 000s of structures typically on a single ESF map. Here, we introduce geometric landscapes, a representation for ESF maps based on geometric similarity, quantified by persistent homology. We show that this representation allows the exploration of complex ESF maps, automatically pinpointing interesting crystalline phases available to the molecule. Furthermore, we show that geometric landscapes can serve as an accountable descriptor for porous materials to predict their performance for gas adsorption applications. A machine learning model trained using this geometric similarity could reach a remarkable accuracy in predicting the materials' performance for methane storage applications.
- This article is part of the themed collections: Celebrating the Chemical Sciences in India - Leaders in the Field Symposium 2020, 2020 Chemical Science HOT Article Collection and Accelerating Chemistry Symposium Collection
 Please wait while we load your content...
Please wait while we load your content...

