
The duality of electron localization and covalency in lanthanide and actinide metallocenes†
 *b
Jason M.
Keith,
c
Stosh A.
Kozimor,
b
Stefan G.
Minasian,
*a
David K.
Shuh,
a
S. Chantal E.
Stieber
*d
and
*b
Jason M.
Keith,
c
Stosh A.
Kozimor,
b
Stefan G.
Minasian,
*a
David K.
Shuh,
a
S. Chantal E.
Stieber
*d
and
Abstract
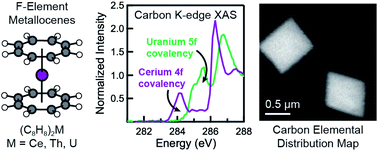
Previous magnetic, spectroscopic, and theoretical studies of cerocene, Ce(C8H8)2, have provided evidence for non-negligible 4f-electron density on Ce and implied that charge transfer from the ligands occurs as a result of covalent bonding. Strong correlations of the localized 4f-electrons to the delocalized ligand π-system result in emergence of Kondo-like behavior and other quantum chemical phenomena that are rarely observed in molecular systems. In this study, Ce(C8H8)2 is analyzed experimentally using carbon K-edge and cerium M5,4-edge X-ray absorption spectroscopies (XAS), and computationally using configuration interaction (CI) calculations and density functional theory (DFT) as well as time-dependent DFT (TDDFT). Both spectroscopic approaches provide strong evidence for ligand → metal electron transfer as a result of Ce 4f and 5d mixing with the occupied C 2p orbitals of the C8H82− ligands. Specifically, the Ce M5,4-edge XAS and CI calculations show that the contribution of the 4f1, or Ce3+, configuration to the ground state of Ce(C8H8)2 is similar to strongly correlated materials such as CeRh3 and significantly larger than observed for other formally Ce4+ compounds including CeO2 and CeCl62−. Pre-edge features in the experimental and TDDFT-simulated C K-edge XAS provide unequivocal evidence for C 2p and Ce 4f covalent orbital mixing in the δ-antibonding orbitals of e2u symmetry, which are the unoccupied counterparts to the occupied, ligand-based δ-bonding e2u orbitals. The C K-edge peak intensities, which can be compared directly to the C 2p and Ce 4f orbital mixing coefficients determined by DFT, show that covalency in Ce(C8H8)2 is comparable in magnitude to values reported previously for U(C8H8)2. An intuitive model is presented to show how similar covalent contributions to the ground state can have different impacts on the overall stability of f-element metallocenes.
 Please wait while we load your content...
Please wait while we load your content...

