
Surface chemistry dictates stability and oxidation state of supported single metal catalyst atoms†

Abstract
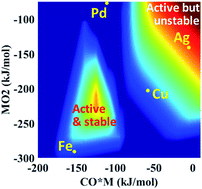
Single atom catalysts receive considerable attention due to reducing noble metal utilization and potentially eliminating certain side reactions. Yet, the rational design of highly reactive and stable single atom catalysts is hampered by the current lack of fundamental insights at the single atom limit. Here, density functional theory calculations are performed for a prototype reaction, namely CO oxidation, over different single metal atoms supported on alumina. The governing reaction mechanisms and scaling relations are identified using microkinetic modeling and principal component analysis, respectively. A large change in the oxophilicity of the supported single metal atom leads to changes in the rate-determining step and the catalyst resting state. Multi-response surfaces are introduced and built cheaply using a descriptor-based, closed form kinetic model to describe simultaneously the activity, stability, and oxidation state of single metal atom catalysts. A double peaked volcano in activity is observed due to competing rate-determining steps and catalytic cycles. Reaction orders of reactants provide excellent kinetic signatures of the catalyst state. Importantly, the surface chemistry determines the stability, oxidation, and resting state of the catalyst.
 Please wait while we load your content...
Please wait while we load your content...

